Sequence Analysis (in Progress)¶
This "Part 1" is a collection of Python-based example codes focussing on sequence analysis on a very basic level (see TOC). It is motivated by the many jobs descriptions currently online for genome analysis, next-gen-sequencing (NGS) advances and my fascination towards life science in general. Apologies for the code often being a little sloppy and "easy-going". Due to a limited time frame, priorities were set to "progress & visualization" instead of "finding the most efficient solution" :)
Whenever a function or method is developed to fulfill the goals, it is outsources into the subfolder "ModulesOwn", take a look and have fun. Check out my website (www.bio-century.net) for demos, literature recommendations, links to data bases and further inspirations.
# export image gallery yes or no
saveGallery = True
# saveGallery = False
Table of Contents¶
- Reference Table
- First Steps in Sequence Analysis (Groundwork)
- Set Up a simple Graphical User Interface (GUI)
- Mutations, Insertions and Deletions
- K-mer-Analysis of a Sequence
- Data Visualization
- Appendix
| Tag or Variable | Meaning |
|---|---|
| mySequence | Target Sequence |
| mySOI | Sequence of Interest |
| ssDNA | single-stranded DNA |
Goal:
- Load a target DNA-sequence ("mySequence") from external file
- split the DNA at first incidence of a dedicated subpart ("mySOI") and
- colorprint a for visualization purposes.
# sources
# - https://www.bioinformatics.org/sms2/random_dna.html
# - https://pkg.go.dev/github.com/whitedevops/colors
# include packages
from ModulesExternal.TerminalColors import TerminalColors
# initialize and define variables and parameter
mySOI="CGCCAAAAA"
# load file line-by-line
with open('./ModulesOwn/A_Groundwork_Data/01_01_SeqOfInterest.txt') as f:
myLines = f.readlines()
# compute and visualize results
# find mySO, split and color it
for ii, mySequence in enumerate(myLines):
SOI_POS = mySequence.upper().rfind(mySOI)
print(mySequence[:SOI_POS - 1]
+ f"{TerminalColors.Green}"
+ mySequence[SOI_POS:SOI_POS + len(mySOI)].upper()
+ f"{TerminalColors.Default}", end = '\n')
print(" "*(SOI_POS - 1)
+ f"{TerminalColors.LightBlue}"
+ mySequence[SOI_POS:SOI_POS + len(mySOI)].upper()
+ f"{TerminalColors.Default}" + mySequence[SOI_POS + 1:], end = '')
ctgggactctagctgatccacccgcctagggcagcacacataggacgtagctCGCCAAAAA CGCCAAAAAgccaaaaagacgaacccaccatgcccagacgcatctggctaagctc
Goal:
To determine all modalities of a DNA-single-strand sequence, i.e.
- the complement-,
- the reverse- as well as
- the reverse-complement-strand
and visualize them.
# sources
# - https://www.bioinformatics.org/sms2/random_dna.html
# include modules
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
from tkinter import *
from tkinter.ttk import *
# initialize and define variables and parameter
mySequenceInput = "ctgggactctagctgatccacccgcctagggcagcacacataggacgtagctgcgccaaaaagacgaacccaccatgcccagacgcatctggctaagctc"
mySequence = mySequenceInput.upper()
myNumberOfSOIs = 3
mySOI = [""] * myNumberOfSOIs
myColors = [""] * myNumberOfSOIs
mySequenceColored = [""] * myNumberOfSOIs
myColorsDNA = [TerminalColors.Green, TerminalColors.Yellow, TerminalColors.Blue, TerminalColors.Magenta]
myDefaultColor = TerminalColors.Default
# compute and visualize results
returnSeq = Groundwork.ComplRev(mySequence,"Sequence")
mySequenceColoredDNA = Groundwork.ColorDNA(returnSeq, myColorsDNA, myDefaultColor)
print("Sequence: \t \t", mySequenceColoredDNA, end = '\n')
print("\t \t \t", ''.join('|' for i in returnSeq), end = '\n')
returnSeq = Groundwork.ComplRev(mySequence,"SequenceComplement")
mySequenceColoredDNA = Groundwork.ColorDNA(returnSeq, myColorsDNA, myDefaultColor)
print("Complement: \t \t", mySequenceColoredDNA, end = '\n')
print("")
returnSeq = Groundwork.ComplRev(mySequence,"SequenceReverse")
mySequenceColoredDNA = Groundwork.ColorDNA(returnSeq, myColorsDNA, myDefaultColor)
print("Reverse: \t \t", mySequenceColoredDNA, end = '\n')
print("\t \t \t", ''.join('|' for i in returnSeq), end = '\n')
returnSeq = Groundwork.ComplRev(mySequence,"SequenceReverseComplement")
mySequenceColoredDNA = Groundwork.ColorDNA(returnSeq, myColorsDNA, myDefaultColor)
print("Reverse Complement: \t", mySequenceColoredDNA, end = '\n')
Sequence: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGGACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Complement: GACCCTGAGATCGACTAGGTGGGCGGATCCCGTCGTGTGTATCCTGCATCGACGCGGTTTTTCTGCTTGGGTGGTACGGGTCTGCGTAGACCGATTCGAG Reverse: CTCGAATCGGTCTACGCAGACCCGTACCACCCAAGCAGAAAAACCGCGTCGATGCAGGATACACACGACGGGATCCGCCCACCTAGTCGATCTCAGGGTC |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Reverse Complement: GAGCTTAGCCAGATGCGTCTGGGCATGGTGGGTTCGTCTTTTTGGCGCAGCTACGTCCTATGTGTGCTGCCCTAGGCGGGTGGATCAGCTAGAGTCCCAG
Goal: To identify and mark the the position of a SOI (mySOI) in a given target sequence (mySequence). The return "positionsSOI" shall be
- 1 if the respective base at position ii of the sequence "mySequence" is part of mySOI
- >1 if two mySOI-regions overlap and
- 0 otherwise.
# sources
# - https://www.bioinformatics.org/sms2/random_dna.html
# include modules
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
# initialize and define variables and parameter
mySequenceInput = "ctgggactctagctgatccacccgcctagggcagcacacataggacgtagctgcgccaaaaagacgaacccaccatgcccagacgcatctggctaagctc"
mySequence = mySequenceInput.upper()
mySOI = "GG"
myCount, mySOIPositions = Groundwork.SOIPositions(mySequence, mySOI, "countchain")
myColor = TerminalColors.Green
myDefaultColor = TerminalColors.Default
mySequenceColored = Groundwork.ColorTheSeq(mySequence, mySOIPositions, myColor, myDefaultColor)
mySOIPositionsColored = Groundwork.ColorTheSeq(mySOIPositions, mySOIPositions, myColor, myDefaultColor)
# compute and visualize results
print("mySequence: \t " + mySequenceColored, end = "\n")
print("mySOIPositions: " + mySOIPositionsColored, end = '\n')
labelNum, labelX = Groundwork.SeqNumberGen(mySequence, 10)
print("\t\t " + labelNum, end = '\n')
print("\t\t " + labelX, end = '\n')
myCount, mySOIPositionsIndices = Groundwork.SOIPositions(mySequence, mySOI, "indices")
print("Indices: \t", mySOIPositionsIndices, end = "\n")
mySequence: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGGACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOIPositions: 0012100000000000000000000000121000000000001100000000000000000000000000000000000000000000001100000000 1234567890123456789012345678901234567890123456789012345678901234567890123456789012345678901234567890 ---------|---------|---------|---------|---------|---------|---------|---------|---------|---------x Indices: [2, 3, 28, 29, 42, 90]
Goal:
To generalize the algorithm above. The positions of 3 given SOIs (mySOIs) shall be
- identified in a target sequence (mySequence),
- labelled individually (green, blue and yellow) and
- a character of the target sequence shall be colored in red, if there is an overlap between the same or different SOIs.
# include modules
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
# initialize and define variables and parameter
dictTargetSequence = {}
dictSOIs = {}
mySequenceInput = "ctgggactctagctgatccacccgcctagggcagcacacataggacgtagctgcgccaaaaagacgaacccaccatgcccagacgcatctggctaagctc"
dictTargetSequence["mySequence"] = mySequenceInput.upper()
dictSOIs["mySOI01"] = "AGG"
dictSOIs["mySOI02"] = "GAC"
dictSOIs["mySOI03"] = "CTAG"
SOICount = [0] * len(dictSOIs)
SOIPositions = [0] * len(dictSOIs)
SOIPositionsIndices = []
mySequenceColored = [""] * len(dictSOIs)
myColors = [TerminalColors.Green, TerminalColors.Blue, TerminalColors.Yellow]
myDefaultColor = TerminalColors.Default
myColorWarning = TerminalColors.Red
dictSOIPositions = {}
dictSOIPositionsIndices = {}
# compute and visualize results
labelNum, labelX = Groundwork.SeqNumberGen(dictTargetSequence["mySequence"], 10)
print("\t\t\t " + labelNum, end = '\n')
print("\t\t\t " + labelX, end = '\n')
for count, value in enumerate(dictSOIs):
SOICount[count], dictSOIPositions[value] = Groundwork.SOIPositions(dictTargetSequence["mySequence"], dictSOIs[value], "countchain")
SOICount[count], dictSOIPositionsIndices[value] = Groundwork.SOIPositions(dictTargetSequence["mySequence"], dictSOIs[value], "indices")
mySequenceColored[count] = Groundwork.ColorTheSeq(dictTargetSequence["mySequence"], dictSOIPositions[value], myColors[count], myDefaultColor)
SOIPositions[count] = dictSOIPositions[value]
print(value + ": \t \t ", mySequenceColored[count], end = '\n')
mySequenceColored, mySOIPositionsTotalColored, mySOIPositionsTotal = Groundwork.ColorTheSeqMerge(dictTargetSequence["mySequence"],
SOIPositions,
myColors,
myDefaultColor,
myColorWarning)
print("-" * 126, end = '\n')
print("mySOIs (total): \t ", mySequenceColored, end = '\n')
print("mySOIPositions (total): ", mySOIPositionsTotalColored, end = '\n')
print("-" * 126, end = '\n')
print("mySOI01 Indices:\t ", dictSOIPositionsIndices["mySOI01"])
print("mySOI02 Indices:\t ", dictSOIPositionsIndices["mySOI02"])
print("mySOI03 Indices:\t ", dictSOIPositionsIndices["mySOI03"])
1234567890123456789012345678901234567890123456789012345678901234567890123456789012345678901234567890 ---------|---------|---------|---------|---------|---------|---------|---------|---------|---------x mySOI01: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGGACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOI02: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGGACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOI03: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGGACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC ------------------------------------------------------------------------------------------------------------------------------ mySOIs (total): CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGGACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOIPositions (total): 0000111011110000000000000112210000000000011211000000000000000011100000000000000001110000000000000000 ------------------------------------------------------------------------------------------------------------------------------ mySOI01 Indices: [27, 41] mySOI02 Indices: [4, 43, 62, 81] mySOI03 Indices: [8, 25]
For further sequence analysis it might be useful to establish data containers in order to group different variables and parameter that belong together. One possibility in Python is to set up a collection (namedtuples). Please keep in mind, that long target sequences should be excluded / stored externally and then should be assigned by a nametag identifyer.
# sources
# - https://stackoverflow.com/questions/9713352/python-creating-a-list-with-a-unique-automatically-generated-name
# - https://www.pythonpool.com/python-string-to-variable-name/
# - https://www.pythonforbeginners.com/basics/convert-string-to-variable-name-in-python
# include modules
from collections import namedtuple
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
# initialize and define variables and parameter
dictTargetSequence = {}
dictSOIs = {}
mySOI = []
dictTargetSequence["mySequence01"] = "ctgggactctagctgatccacccgcctagggcagcacacatagg".upper()
dictTargetSequence["mySequence02"] = "acgtagctgcgccaaaaagacgaacccaccatgcccagacgcatctggctaagctc".upper()
dictSOIs["mySOI01"] = "AGG"
dictSOIs["mySOI02"] = "GAC"
dictSOIs["mySOI03"] = "CTAG"
SOICount = [0] * len(dictSOIs)
SOIPositions = [0] * len(dictSOIs)
dictIndices = {}
mySequenceColored = [""] * len(dictSOIs)
myColors = [TerminalColors.Green, TerminalColors.Blue, TerminalColors.Yellow]
myColorsStr = ["Green", "Blue", "Yellow","Green", "Blue", "Yellow"] # Terminal Colors are tricky to store. Using identifiers instead
myDefaultColor = TerminalColors.Default
myColorWarning = TerminalColors.Red
mySOIsAndIndices = namedtuple("myDataCollection","mySequenceKey mySequenceValue mySOIKey mySOIValue myColors myIndices")
mySOIsAndIndices_array = []
mySeqPos = []
dictSOIPositions = {}
dictSOIPositionsIndices = {}
# compute and visualize results
for mySeqsKey in dictTargetSequence:
kk = 0
for kk, myCurrentKey in enumerate(dictSOIs):
mySOIsAndIndices_array.append(mySOIsAndIndices(mySeqsKey, dictTargetSequence[mySeqsKey], myCurrentKey, dictSOIs[myCurrentKey], myColorsStr[kk], []))
for countTargetSeq, valueTargetSeq in enumerate(dictTargetSequence):
print(valueTargetSeq)
labelNum, labelX = Groundwork.SeqNumberGen(dictTargetSequence[valueTargetSeq], 10)
print("\t\t\t " + labelNum, end = '\n')
print("\t\t\t " + labelX, end = '\n')
for count, value in enumerate(dictSOIs):
SOICount[count], dictSOIPositions[value] = Groundwork.SOIPositions(dictTargetSequence[valueTargetSeq], dictSOIs[value], "countchain")
SOICount[count], dictSOIPositionsIndices[value] = Groundwork.SOIPositions(dictTargetSequence[valueTargetSeq], dictSOIs[value], "indices")
mySequenceColored[count] = Groundwork.ColorTheSeq(dictTargetSequence[valueTargetSeq], dictSOIPositions[value], myColors[count], myDefaultColor)
SOIPositions[count] = dictSOIPositions[value]
print(value + ": \t \t ", mySequenceColored[count], end = '\n')
print("\n")
for SOIs in mySOIsAndIndices_array:
for ii in range(len(SOIs.mySequenceValue) - len(SOIs.mySOIValue)):
if SOIs.mySequenceValue[ii:ii + len(SOIs.mySOIValue)] == SOIs.mySOIValue:
SOIs.myIndices.append(ii)
print(SOIs)
mySequence01 12345678901234567890123456789012345678901234 ---------|---------|---------|---------|---- mySOI01: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG mySOI02: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG mySOI03: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG mySequence02 12345678901234567890123456789012345678901234567890123456 ---------|---------|---------|---------|---------|------ mySOI01: ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOI02: ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOI03: ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC myDataCollection(mySequenceKey='mySequence01', mySequenceValue='CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG', mySOIKey='mySOI01', mySOIValue='AGG', myColors='Green', myIndices=[27]) myDataCollection(mySequenceKey='mySequence01', mySequenceValue='CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG', mySOIKey='mySOI02', mySOIValue='GAC', myColors='Blue', myIndices=[4]) myDataCollection(mySequenceKey='mySequence01', mySequenceValue='CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG', mySOIKey='mySOI03', mySOIValue='CTAG', myColors='Yellow', myIndices=[8, 25]) myDataCollection(mySequenceKey='mySequence02', mySequenceValue='ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC', mySOIKey='mySOI01', mySOIValue='AGG', myColors='Green', myIndices=[]) myDataCollection(mySequenceKey='mySequence02', mySequenceValue='ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC', mySOIKey='mySOI02', mySOIValue='GAC', myColors='Blue', myIndices=[18, 37]) myDataCollection(mySequenceKey='mySequence02', mySequenceValue='ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC', mySOIKey='mySOI03', mySOIValue='CTAG', myColors='Yellow', myIndices=[])
The accessibility to the info here stored as values can be a little tricky though. Here are two possibilities to do that:
# initialize and define variables and parameter
# TargetSeq SOI Item2Look4
SEARCH_ITEM=["mySequence02", "mySOI02", "myColors"]
# compute and visualize results
INDEX_ARRAY=(list(dictTargetSequence.keys()).index(SEARCH_ITEM[0])) * len(dictSOIs) + (list(dictSOIs.keys()).index(SEARCH_ITEM[1]))
print("You are asking for the value of \"" + SEARCH_ITEM[2] + "\" for the sequence of interest \"" + SEARCH_ITEM[1] + "\" within the target sequence \"" + SEARCH_ITEM[0] + "\".")
print("-" * 124, end = '\n')
res = mySOIsAndIndices_array.__getitem__(INDEX_ARRAY).__getitem__(mySOIsAndIndices._fields.index(SEARCH_ITEM[2]))
print("The value is as follows: \t ", res)
You are asking for the value of "myColors" for the sequence of interest "mySOI02" within the target sequence "mySequence02". ---------------------------------------------------------------------------------------------------------------------------- The value is as follows: Blue
# initialize and define variables and parameter
# Data-pair2look4:
# key value
SEARCH_ITEM=["mySOIKey", "mySOI02"]
# compute and visualize results
print("All items fulfilling the requirements:")
print("-" * 197, end = '\n')
for SOIs in mySOIsAndIndices_array:
test_data = SOIs.__getitem__(mySOIsAndIndices._fields.index(SEARCH_ITEM[0]))
if test_data == SEARCH_ITEM[1]:
res = SOIs.__getitem__(mySOIsAndIndices._fields.index(SEARCH_ITEM[0]))
print(SOIs)
All items fulfilling the requirements: ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- myDataCollection(mySequenceKey='mySequence01', mySequenceValue='CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG', mySOIKey='mySOI02', mySOIValue='GAC', myColors='Blue', myIndices=[4]) myDataCollection(mySequenceKey='mySequence02', mySequenceValue='ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC', mySOIKey='mySOI02', mySOIValue='GAC', myColors='Blue', myIndices=[18, 37])
1.5.2 Store in JSON-Format¶
Probably more convient, common and thus advisory is to use a dictionary / JSON-format combination.
Step 1: Dicitonary Setup and Test-Readout
# sources
# - https://python.plainenglish.io/how-to-read-and-write-to-json-file-in-python-ef35460aaeb5
# initialize and define variables and parameter
# Dicitonary Setup
dictSeqGroup = {
"SOIs": {
"mySOI01": {
"Seq": "AGG",
"myColor": "Green",
},
"mySOI02": {
"Seq": "GAC",
"myColor": "Blue",
},
"mySOI03": {
"Seq": "CTAG",
"myColor": "Yellow",
}
},
"targetSeq": {
"mySequence01": "CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG",
"mySequence02": "ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC"
}
}
# compute and visualize results
# Test-Readout
print(dictSeqGroup["targetSeq"]["mySequence02"]) # <-- 1. query
print(dictSeqGroup["SOIs"]["mySOI03"]["myColor"]) # <-- 2. query
ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC Yellow
Step 2: Data Export into JSON-Format
# sources
# - https://pynative.com/python-prettyprint-json-data/
# - https://stackoverflow.com/questions/73800772/returning-jsondecodeerror-expecting-value
# include modules
import json
with open("./ModulesOwn/A_Groundwork_Data/01_05_02_DictSeqGroup.json", "w") as outfile:
json.dump(dictSeqGroup, outfile, indent = 4, sort_keys = True)
Step 3: Run Operations on Data and Save Modifications. Here: We determine and add the SOIPositions-Indices to each tarSeq-mySOI-pair, include the data in the master-dictionary and save everything in JSON-format.
# initialize and define variables and parameter
myColors = [TerminalColors.Green, TerminalColors.Blue, TerminalColors.Yellow]
myDefaultColor = TerminalColors.Default
myColorWarning = TerminalColors.Red
# (Re-)Open JSON file
with open('./ModulesOwn/A_Groundwork_Data/01_05_02_DictSeqGroup.json', 'r') as openfile:
# Reading from json file
dictSeqGroup = json.load(openfile)
# compute and visualize results
# Determine and print indices in terminal
for countTargetSeq, valueTargetSeq in enumerate(dictSeqGroup["targetSeq"].keys()):
labelNum, labelX = Groundwork.SeqNumberGen(dictSeqGroup["targetSeq"][valueTargetSeq], 10)
print("\t\t\t " + labelNum, end = '\n')
print("\t\t\t " + labelX, end = '\n')
for count, value in enumerate(dictSeqGroup["SOIs"].keys()):
SOICount[count], dictSOIPositions[value] = Groundwork.SOIPositions(dictSeqGroup["targetSeq"][valueTargetSeq], dictSeqGroup["SOIs"][value]["Seq"], "countchain")
SOICount[count], dictSOIPositionsIndices[value] = Groundwork.SOIPositions(dictSeqGroup["targetSeq"][valueTargetSeq], dictSeqGroup["SOIs"][value]["Seq"], "indices")
mySequenceColored[count] = Groundwork.ColorTheSeq(dictSeqGroup["targetSeq"][valueTargetSeq], dictSOIPositions[value], myColors[count], myDefaultColor)
SOIPositions[count] = dictSOIPositions[value]
print(value + ": \t \t ", mySequenceColored[count], end = '\n')
if "myIndices" in dictSeqGroup["SOIs"][value]:
pass
else:
dictSeqGroup["SOIs"][value]["myIndices"] = {}
dictSeqGroup["SOIs"][value]["myIndices"][valueTargetSeq] = dictSOIPositionsIndices[value]
for count, value in enumerate(dictSeqGroup["SOIs"].keys()):
print("\t\t\t ", dictSOIPositionsIndices[value], end = '\n')
print("\n")
# Write info into json file
with open("./ModulesOwn/A_Groundwork_Data/01_05_02_DictSeqGroup.json", "w") as outfile:
json.dump(dictSeqGroup, outfile, indent = 4, sort_keys = True)
12345678901234567890123456789012345678901234 ---------|---------|---------|---------|---- mySOI01: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG mySOI02: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG mySOI03: CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG [27, 41] [4] [8, 25] 12345678901234567890123456789012345678901234567890123456 ---------|---------|---------|---------|---------|------ mySOI01: ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOI02: ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC mySOI03: ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC [] [18, 37] []
Step 4: Reload JSON-file and print the content. The indices are now added to each targetSeq-mySOI-pair.
# sources
# - https://www.geeksforgeeks.org/read-a-file-line-by-line-in-python/
# Using readlines()
file1 = open('./ModulesOwn/A_Groundwork_Data/01_05_02_DictSeqGroup.json', 'r')
Lines = file1.readlines()
# compute and visualize results
count = 0
# Strips the newline character
for line in Lines:
count += 1
print(line, end = '')
{
"SOIs": {
"mySOI01": {
"Seq": "AGG",
"myColor": "Green",
"myIndices": {
"mySequence01": [
27,
41
],
"mySequence02": []
}
},
"mySOI02": {
"Seq": "GAC",
"myColor": "Blue",
"myIndices": {
"mySequence01": [
4
],
"mySequence02": [
18,
37
]
}
},
"mySOI03": {
"Seq": "CTAG",
"myColor": "Yellow",
"myIndices": {
"mySequence01": [
8,
25
],
"mySequence02": []
}
}
},
"targetSeq": {
"mySequence01": "CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACACATAGG",
"mySequence02": "ACGTAGCTGCGCCAAAAAGACGAACCCACCATGCCCAGACGCATCTGGCTAAGCTC"
}
}
Goal:
Set up a simple first draft of a Grahical User Interface (GUI) for future applications. The GUI should
- analyse a given DNA-Sequence with respect to 3 mySOIs and color code the respective positions in the target sequence
- save the results in an external file
- look fancy
- provide space for a next analysis step. Prefered is a tab-design of the GUI.
# sources
# - https://www.delftstack.com/howto/python-tkinter/tkinter-tabs/
# - https://github.com/rdbende/Azure-ttk-theme
# - https://www.javatpoint.com/python-tkinter-text
# - https://learn.microsoft.com/en-us/windows/win32/api/dwmapi/ne-dwmapi-dwmwindowattribute
# - https://www.plus2net.com/python/tkinter-grid.php
# - https://www.pythontutorial.net/tkinter/tkinter-validation/
# - https://stackoverflow.com/questions/17251016/python-tkinter-how-to-change-the-windows-border-color
# - https://stackoverflow.com/questions/18052395/array-of-buttons-in-python
# - https://stackoverflow.com/questions/53461066/python-showing-next-tab-by-pressing-a-button
# - https://stackoverflow.com/questions/70224682/is-there-a-way-in-tkinter-to-left-align-tabs-notebook
# - https://www.tutorialspoint.com/how-to-save-the-contents-of-a-textbox-in-tkinter
# - https://www.tutorialspoint.com/how-to-change-the-color-of-certain-words-in-a-tkinter-text-widget
# include modules
from IPython.display import Image as ImageIPython
import json
import TKinterModernThemes as TKMT
import ModulesOwn.A_Groundwork as Groundwork
import ModulesOwn.B_SimpleTabbedGUI as SimpleTabbedGUI
from ModulesExternal.TerminalColors import TerminalColors
import numpy as np
from PIL import Image, ImageTk
import sys
from tkinter import Text, END
from tkinter import *
from tkinter.ttk import *
import tkinter as tk
from tkinter import ttk
def main():
# convert BCN-image to .ico in order to make image applicable as a logo for the window frame
from PIL import Image
logo = Image.open("./icons/logoBCNicon.png")
logo.save("./icons/logoBCNicon.ico", format = 'ICO')
# define colors and constants
myForegroundColor = "#4c87c8"
myBackgroundColor = "#000a34"
dictSequence = {}
mySOI = []
myNumberLabelBlank = 30
myLabelBlank = [""] * myNumberLabelBlank
myDefaultColor = TerminalColors.Default
myColorWarning = TerminalColors.Red
myDefaultColorTextBox = "white"
myColorWarningTextBox = "red"
myInput = [""] * 4
myColorsTextBox = []
myColors = [TerminalColors.Green, TerminalColors.Blue, TerminalColors.Yellow]
# initialize GUI
root = tk.Tk()
# set graphical appearance
root.configure(background = myBackgroundColor)
Groundwork.DarkTitleBar(root)
root.title('Tabbed GUI')
root.tk.call("source", "./_themes/Sun-Valley-ttk-theme-main/sv.tcl")
root.tk.call("set_theme", "dark")
root.iconbitmap("./icons/logoBCNicon.ico")
ttk.Style().configure("RB.TButton", foreground = myForegroundColor)
# create a tab control that manages multiple tabs
tabsystem = ttk.Notebook(root)
# create new tabs using frame widget
tab1 = Frame(tabsystem)
tab2 = Frame(tabsystem)
# initialize some blank labels used for GUI design
for ii in range(0, len(myLabelBlank)):
myLabelBlank[ii] = ttk.Label(tab1, text = " ", background = myBackgroundColor)
# set style
s = ttk.Style(root)
s.configure("TFrame", background = myBackgroundColor, foreground = myBackgroundColor)
s.configure("TNotebook", background = myBackgroundColor, foreground = myBackgroundColor)
s.configure("TNotebook.Tab", background = myBackgroundColor, foreground = "white")
# load (previous) dataset
with open("./ModulesOwn/B_SimpleTabbedGUI_Data/02_Sequence.json", "r") as openfile:
dictSequence = json.load(openfile)
for ii in range(len(dictSequence["SOIs"])):
myColorsTextBox.append(dictSequence["SOIs"]["mySOI""{:02d}".format(ii+1)]["myColor"])
# set top label
columnCount = 0; blankCount = 0
myLabelBlank[blankCount].grid(row = columnCount, column = 0); blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 2); blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 4); blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 6); columnCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 8); columnCount += 1
myLabel = tk.Label(tab1, text = "Sequence Analysis", font = ('Arial', 14, 'bold'), bg = myBackgroundColor)
myLabel.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E)
columnCount += 1
blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0)
myValidation = [] * 4
# set Input textboxes and labels
for ii in range(len(dictSequence["SOIs"]) + 1):
blankCount += 1
myLabelBlank[blankCount+ii].grid(row = columnCount, column = 2)
columnCount += 1
if ii == 0:
currentKey = "mySequence"
currentValue = dictSequence["mySequence"]
myLabel = ttk.Label(tab1, text = currentKey + ": ", anchor="e", justify = "left",
font = ('Courier New', 12, 'bold'),
foreground = "white", background = myBackgroundColor)
myLabel.grid(row = columnCount, column = 1, sticky = tk.W + tk.E)
myInput[ii] = ttk.Entry(tab1, width = 40, style = "EntryStyle.TEntry",
font = ('Courier New', 12, 'bold'),
foreground = myForegroundColor, background = myBackgroundColor)
myInput[ii].insert("end", currentValue)
else:
currentKey = "mySOI""{:02d}".format(ii)
currentValue = dictSequence["SOIs"][currentKey]["Seq"]
myLabel = ttk.Label(tab1, text = currentKey + ": ", anchor = "e", justify = "left",
font = ('Courier New', 12, 'bold'), background = myBackgroundColor,
foreground = myColorsTextBox[ii - 1])
myLabel.grid(row = columnCount, column = 1, sticky = tk.W + tk.E)
myInput[ii] = ttk.Entry(tab1, width = 40, style = "EntryStyle.TEntry",
font = ('Courier New', 12, 'bold'),
foreground = myForegroundColor, background = myBackgroundColor)
myInput[ii].insert("end", currentValue)
myInput[ii].grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5)
blankCount += 1; columnCount += 1
myLabelBlank[blankCount + ii].grid(row = columnCount, column = 2)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
# determine colored result for textfield of GUI
myNumberOfSOIs = len(dictSequence["SOIs"])
myCount = [0] * myNumberOfSOIs
mySOIPositions = [0] * myNumberOfSOIs
mySequenceColored = [""] * myNumberOfSOIs
mySequence = dictSequence["mySequence"]
columnCount += 1
for ii in range(len(dictSequence["SOIs"])):
currentKey = "mySOI""{:02d}".format(ii + 1)
currentValue = dictSequence["SOIs"][currentKey]["Seq"]
myCount[ii], mySOIPositions[ii] = Groundwork.SOIPositions(mySequence, currentValue, "countchain")
mySequenceColored[ii] = Groundwork.ColorTheSeq(mySequence, mySOIPositions[ii], myColors[ii], myDefaultColor)
mySequenceColored, mySOIPositionsTotalColored, mySOIPositionsTotal = Groundwork.ColorTheSeqMerge(mySequence, mySOIPositions, myColors,
myDefaultColor, myColorWarning)
print("mySOIs (total): \t \t", mySequenceColored, end = '\n')
print("mySOIPositions (total) : \t", mySOIPositionsTotalColored, end = '\n')
myLabel = ttk.Label(text = mySequence, font=('Arial', 14, 'bold'))
# write colored result in textfield of GUI
myText = Text(tab1, height = 3, width = 35, background = myBackgroundColor, highlightthickness = 0,
borderwidth = 0, font = ('Courier New', 12, 'bold'))
SimpleTabbedGUI.AnalyseSeq(myText, myNumberOfSOIs, mySequence, mySOIPositions, myColorsTextBox,
myDefaultColorTextBox, myColorWarningTextBox, myBackgroundColor, tab1, columnCount, myInput)
myText.delete
myTextnew= Text(tab1, height = 2, width = 35, background = myBackgroundColor, highlightthickness = 0,
borderwidth = 0, font = ('Courier New', 12, 'bold'))
myTextnew = SimpleTabbedGUI.ColorTheSeqMergeGUI(myTextnew, dictSequence["mySequence"], mySOIPositions, myColorsTextBox,
myDefaultColorTextBox, myColorWarningTextBox)
myTextnew.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E)
myColumnCountAnalysisStatic = columnCount; columnCount += 1
# imprint user manual
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
myLabelLegend = ttk.Label(tab1, text = "Sequences of Interest (SOIs): Green, blue and yellow", anchor = "w", justify = "left",
font = ('Courier New', 10, 'bold'), background = myBackgroundColor, foreground = "white")
myLabelLegend.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E); columnCount += 1
myLabelLegend = ttk.Label(tab1, text = "Overlaps: red", anchor = "w", justify = "left", font = ('Courier New', 10, 'bold'),
background = myBackgroundColor, foreground = "white")
myLabelLegend.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E)
columnCount += 1; columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
myLabelMan = ttk.Label(tab1,text = "Manual: ", anchor = "w", justify = "left", font = ('Courier New', 10, 'bold'),
background = myBackgroundColor, foreground = "white")
myLabelMan.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E); columnCount += 1
myLabelMan = ttk.Label(tab1,text = "1. Modify Sequences", anchor = "w", justify="left", font = ('Courier New', 10, 'bold'),
background = myBackgroundColor, foreground = "white")
myLabelMan.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E); columnCount += 1
myLabelMan = ttk.Label(tab1,text = "2. Click \"Analyze\"", anchor = "w", justify = "left", font = ('Courier New', 10, 'bold'),
background = myBackgroundColor, foreground = "white")
myLabelMan.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E); columnCount += 1
myLabelMan = ttk.Label(tab1,text = "3. Click \"Save\"", anchor = "w", justify = "left", font = ('Courier New', 10, 'bold'),
background = myBackgroundColor, foreground = "white")
myLabelMan.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5, sticky = tk.W + tk.E)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
# Set control buttons and define actions
myButtonAnalysis = ttk.Button(tab1,text = "Analyze", style = "RB.TButton", command = lambda: SimpleTabbedGUI.AnalyseSeq(myText, myNumberOfSOIs,
mySequence, mySOIPositions,
myColorsTextBox, myDefaultColorTextBox,
myColorWarningTextBox, myBackgroundColor,
tab1, myColumnCountAnalysisStatic, myInput))
myButtonAnalysis.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 3, sticky = tk.W + tk.E)
myButtonSave = ttk.Button(tab1, text = "Save", style = "RB.TButton", command = lambda: SimpleTabbedGUI.SaveSequence(dictSequence, myInput))
myButtonSave.grid(row = columnCount, column = 7, sticky = tk.W + tk.E)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
myButton = ttk.Button(tab1, text = "Restore", style = "RB.TButton", command = lambda: SimpleTabbedGUI.LoadDataset(myInput))
myButton.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 3, sticky = tk.W + tk.E)
myButtonClose = ttk.Button(tab1, text = "Close", style = "RB.TButton", command = root.destroy)
myButtonClose.grid(row = columnCount, column = 7, sticky = tk.W + tk.E)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
myButtonClear = ttk.Button(tab1, text = "Clear", style = "RB.TButton", command = lambda: SimpleTabbedGUI.ClearTextandFields(dictSequence, myInput))
myButtonClear.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 3, sticky = tk.W + tk.E)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
# set logo bio-century.net
myImage = Image.open('./icons/logoBCN.png')
imageResized = myImage.resize((288, 47))
myIcon=ImageTk.PhotoImage(imageResized)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0)
myLogoLabel = ttk.Label(tab1, image = myIcon, padding = 5, background = myBackgroundColor)
myLogoLabel.grid(row = columnCount, column = 3, rowspan = 1, columnspan = 5)
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
myButtonNext = ttk.Button(tab1, text="Next Tab", command = lambda: tabsystem.select(tab2))
myButtonNext.grid(row = columnCount, column=7, sticky = tk.W + tk.E); columnCount += 1
columnCount += 1; blankCount += 1
myLabelBlank[blankCount].grid(row = columnCount, column = 0); columnCount += 1
# set "Next Idea" tab
label2nd = Label(tab2, text = "Here is some space for a new idea!", background = myBackgroundColor)
label2nd.grid(column = 1, row = 1, padx = 170,pady = 250)
tabsystem.add(tab1, text = 'Sequence Analysis')
tabsystem.add(tab2, text = 'Next Idea')
tabsystem.grid()
display(ImageIPython(filename = './figures/TabbedGUI.png'))
root.mainloop()
if __name__ == "__main__":
main()
mySOIs (total): CTGGGACTCTAGCTGATCCACCCGCCTAGGGCAGCACA mySOIPositions (total) : 00001110111100000000000001122100000000

3.1 Point Mutations¶
3.1.1 Hamming Distance¶
# include modules
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
import ModulesOwn.C_Mutations as Mutations
# initialize and define variables and parameter
dictTargetSequence = {}
# 123 456 789 012 345 678 901 234 567 890
# M L G L W L L A D *
dictTargetSequence["mySeq01"] = "atg ctg gga ctc tgg ctg ctg gct gat taa".replace(" ","").upper()
dictTargetSequence["mySeq02"] = "atg ctg gTa ctc tAA ctA ctg gAt gat taa".replace(" ","").upper()
# compute hamming distance
hammingDistance, mismatchDNA = Mutations.hammingDistance(dictTargetSequence["mySeq01"], dictTargetSequence["mySeq02"])
# print results
print("-" * 50)
print(" " * 5 + "Results Comparison mySeq01 And mySeq02")
print("-" * 50)
print(" " * 2 + "Hamming Distance: \t" + str(hammingDistance))
print(" " * 2 + "At Position: \t \t" + str(mismatchDNA))
print("-" * 50)
print()
Mutations.printSeqDifferences(dictTargetSequence["mySeq01"], dictTargetSequence["mySeq02"], mismatchDNA)
--------------------------------------------------
Results Comparison mySeq01 And mySeq02
--------------------------------------------------
Hamming Distance: 5
At Position: [7, 13, 14, 17, 22]
--------------------------------------------------
mySeq01: ATGCTGGGACTCTGGCTGCTGGCTGATTAA
||||||| ||||| || |||| |||||||
mySeq02: ATGCTGGTACTCTAACTACTGGATGATTAA
3.1.2 Classify Mutations¶
# sources
# - http://biopython.org/DIST/docs/tutorial/Tutorial.html
# data source
# - https://www.ncbi.nlm.nih.gov/datasets/taxonomy/2697049/
# Severe acute respiratory syndrome coronavirus 2
# include modules
from Bio.Seq import Seq
import ModulesOwn.C_Mutations as Mutations
dictTargetSequence["mySeq01Translated"] = Seq(dictTargetSequence["mySeq01"]).translate()
dictTargetSequence["mySeq02Translated"] = Seq(dictTargetSequence["mySeq02"]).translate()
hammingDistance, mismatchAA = Mutations.hammingDistance(dictTargetSequence["mySeq01Translated"], dictTargetSequence["mySeq02Translated"])
labelNum, labelX = Groundwork.SeqNumberGen(dictTargetSequence["mySeq01Translated"], 10)
print("\t\t" + labelNum, end = '\n')
Mutations.printSeqDifferences(dictTargetSequence["mySeq01Translated"], dictTargetSequence["mySeq02Translated"], mismatchAA)
print("\n")
# print results
print("-" * 50)
print(" " * 5 + "Results Comparison mySeq01 And mySeq02")
print("-" * 50)
print(" " * 2 + "Hamming Distance: \t" + str(hammingDistance))
print(" " * 2 + "At (Position -1): \t" + str(mismatchAA))
print("-" * 50)
print("")
Mutations.printMutationCharakterAminoAcidSeqs(dictTargetSequence["mySeq01"], dictTargetSequence["mySeq02"], mismatchDNA)
1234567890 mySeq01: MLGLWLLAD* || | || || mySeq02: MLVL*LLDD* -------------------------------------------------- Results Comparison mySeq01 And mySeq02 -------------------------------------------------- Hamming Distance: 3 At (Position -1): [2, 4, 7] -------------------------------------------------- G -> V at position 3: Missense Conservative Mutation W -> * at position 5: Nonsense Mutation L -> L at position 6: Silent Mutation A -> D at position 8: Missense Non-Conservative Mutation
Goal:
Create a DNA map depicting the genes of Sars-CoV-2 reversly-transcribed genome (Sars-CoV-2 has an RNA-genome). Show a close-up of the relatively small envelope protein E.
Step 01: Load in genome from fna-file. Show DNA-sequence and the translated result.
# sources:
# https://www.nature.com/articles/s41392-021-00653-w/figures/1
# https://datagy.io/python-find-index-substring/
# - https://en.wikipedia.org/wiki/Conservative_replacement
# data source:
# https://www.ncbi.nlm.nih.gov/datasets/taxonomy/2697049/
# include modules
import re
from Bio.Seq import Seq
from ModulesExternal.TerminalColors import TerminalColors
import warnings
warnings.filterwarnings('ignore')
def findGenes(mySeq, startPos, endPos):
mySeqColoredList = []
mySeqTmpTranslatedIndicesM = []
mySeqTmpTranslatedIndicesStars = []
for ii in range(3):
mySeqTmp = mySeq[startPos + ii : endPos]
mySeqTmpTranslated = Seq(mySeqTmp).translate()
if mySeqTmpTranslated[0] == "M" and mySeqTmpTranslated[-1] == "*":
mySeqColored = TerminalColors.Green + mySeqTmpTranslated + TerminalColors.Default
else:
mySeqColored = TerminalColors.Red + mySeqTmpTranslated + TerminalColors.Default
mySeqColoredList.append(mySeqColored)
indices_object = re.finditer(pattern='M', string = str(mySeqTmpTranslated))
mySeqTmpTranslatedIndicesM.append([index.start() for index in indices_object])
indices_object = re.finditer(pattern='\*', string = str(mySeqTmpTranslated))
mySeqTmpTranslatedIndicesStars.append([index.start() for index in indices_object])
return mySeqColoredList, mySeqTmpTranslatedIndicesM, mySeqTmpTranslatedIndicesStars
# initialize and define variables and parameter
dictGenes = {}
counterGenes = 0
mySeq = ""
myPositioningFile = open('./Data/cds.fna', 'r')
mySeqFile = open('./Data/genomic.fna', 'r')
lines = myPositioningFile.readlines()
for ii, line in enumerate(lines):
if line.find(">NC_045512.2:") > -1:
lineCropped = line[len(">NC_045512.2:"):line.index("[") - 1]
position = lineCropped[:lineCropped.index(" ")]
positionStart = position[:position.index("-")]
positionEnd = position[position.index("-")+1:]
geneName = lineCropped[lineCropped.index(" ") + 1:]
dictGenes[counterGenes] = {}
dictGenes[counterGenes]["Name"] = geneName
dictGenes[counterGenes]["positionStart"] = positionStart
dictGenes[counterGenes]["positionEnd"] = positionEnd
counterGenes += 1
myPositioningFile.close()
lines = mySeqFile.readlines()
for ii, line in enumerate(lines):
if ii > 0:
mySeq = mySeq + line
mySeqFile.close()
mySeq = mySeq.replace("\n","")
# protein E envelope: print all three reading frames
startPos = 26242
endPos = 26472
mySeqColoredList, mySeqIndicesM, mySeqIndicesStars = findGenes(mySeq, startPos, endPos)
dashlineLength = 101
indentHeadlines = 45
ii = 0
print("-" * dashlineLength)
print(" " * indentHeadlines + "Reading Frame 01".upper())
print("-" * dashlineLength)
print("Sequence translated: \t " + str(mySeqColoredList[ii]), end = '\n')
print("Gene Start Position: \t " + str(mySeqIndicesM[ii]), end = '\n')
print("Gene Stop Position: \t " + str(mySeqIndicesStars[ii]), end = '\n')
print("-" * dashlineLength)
print("")
ii += 1
print("-" * dashlineLength)
print(" " * indentHeadlines + "Reading Frame 02".upper())
print("-" * dashlineLength)
print("Sequence translated: \t " + str(mySeqColoredList[ii]), end = '\n')
print("Gene Start Position: \t " + str(mySeqIndicesM[ii]), end = '\n')
print("Gene Stop Position: \t " + str(mySeqIndicesStars[ii]), end = '\n')
print("-" * dashlineLength)
print("")
ii += 1
print("-" * dashlineLength)
print(" " * indentHeadlines + "Reading Frame 03".upper())
print("-" * dashlineLength)
print("Sequence translated: \t " + str(mySeqColoredList[ii]), end = '\n')
print("Gene Start Position: \t " + str(mySeqIndicesM[ii]), end = '\n')
print("Gene Stop Position: \t " + str(mySeqIndicesStars[ii]), end = '\n')
print("-" * dashlineLength)
-----------------------------------------------------------------------------------------------------
READING FRAME 01
-----------------------------------------------------------------------------------------------------
Sequence translated: LCTHSFRKRQVR**LIAYFFFLLSWYSC*LH*PSLLRFDCVRTAAILLT*VL*NLLFTFTLVLKI*ILLEFLIFWS
Gene Start Position: []
Gene Stop Position: [12, 13, 28, 31, 49, 52, 65]
-----------------------------------------------------------------------------------------------------
-----------------------------------------------------------------------------------------------------
READING FRAME 02
-----------------------------------------------------------------------------------------------------
Sequence translated: YVLIRFGRDRYVNS**RTSFSCFRGILASYTSHPYCASIVCVLLQYC*RESCKTFFLRLLSC*KSEFF*SS*SSGL
Gene Start Position: []
Gene Stop Position: [14, 15, 47, 62, 68, 71]
-----------------------------------------------------------------------------------------------------
-----------------------------------------------------------------------------------------------------
READING FRAME 03
-----------------------------------------------------------------------------------------------------
Sequence translated: MYSFVSEETGTLIVNSVLLFLAFVVFLLVTLAILTALRLCAYCCNIVNVSLVKPSFYVYSRVKNLNSSRVPDLLV*
Gene Start Position: [0]
Gene Stop Position: [75]
-----------------------------------------------------------------------------------------------------
# sources:
# https://github.com/Edinburgh-Genome-Foundry/DnaFeaturesViewer/blob/master/examples/overview_and_detail.py
# https://github.com/Edinburgh-Genome-Foundry/DnaFeaturesViewer
# include modules
from dna_features_viewer import GraphicFeature, GraphicRecord
from dna_features_viewer import GraphicFeature, GraphicRecord
import matplotlib.pyplot as plt
featuresList = []
whitelist = [0,1,2,3,4,5,7,8,9,10,11,12,13,14,17,18,19,20,21,22,23,24,25,26,27]
for ii in whitelist: #range(len(dictGenes)):
if ii == 14:
featuresList.append(GraphicFeature(start = int(dictGenes[ii]["positionStart"]), end = int(dictGenes[ii]["positionEnd"]), strand =+ 0, color = "#ffaaaa", label = dictGenes[ii]["Name"], alpha=0.0))
else:
featuresList.append(GraphicFeature(start = int(dictGenes[ii]["positionStart"]), end = int(dictGenes[ii]["positionEnd"]), strand =+ 0, color = "#ccccff", label = dictGenes[ii]["Name"], alpha=0.15))
record = GraphicRecord(sequence = mySeq,features=featuresList)
# record = GraphicRecord(sequence_length=32000,features=featuresList)
# ax1, _ = record.plot(figsize=(50, 2))#figure_width = 14)
# fig, (ax1) = plt.subplots(1, 1, figsize=(50, 2))
ax1, _ = record.plot(figure_width = 40)
ax1.set_title("Sequence detail", loc='left', weight='bold')
ax1.fill_between((startPos, endPos), +1000, -1000, color="#ccffcc", alpha=0.65)
fig, (ax2) = plt.subplots(1, 1, figsize=(40, 2))
# fig, (ax2) = plt.subplots(1, 1, figure_width = 14)
zoom_start, zoom_end = 398, 428 # coordinates of the "detail"
cropped_record = record.crop((startPos-4, endPos+2))
cropped_record.plot_translation(ax=ax2, location=(startPos-4, endPos+2), fontdict={'weight': 'bold'})
cropped_record.plot(ax=ax2, plot_sequence=True)
ax2.set_title("Sequence detail", loc='left', weight='bold')
ax1.figure.savefig('./Figures/sequence_and_translation.png', bbox_inches='tight')
if saveGallery == True:
ax1.figure.savefig('./../GALLERY/Sequence Analysis/03_01_03_Gene_Map01.png', bbox_inches='tight')
ax2.figure.savefig('./../GALLERY/Sequence Analysis/03_01_03_Gene_Map02.png', bbox_inches='tight')
plt.show()


# https://stackoverflow.com/questions/68140575/styling-the-background-color-of-pandas-index-cell
# https://stackoverflow.com/questions/6667201/how-to-define-a-two-dimensional-array
# https://www.youtube.com/watch?v=of3B02hZGS0&t=1583s
# https://www.youtube.com/watch?v=b6xBvl0yPAY
# include modules
import numpy as np
import pandas as pd
# initialize and define variables and parameter
dictTargetSequence = {}
# dictTargetSequence["mySeq01"] = "tgg tg".replace(" ","").upper() # x
# dictTargetSequence["mySeq02"] = "atc gt".replace(" ","").upper() # y
# scoringWeight = [1, -1, -2]
# dictTargetSequence["mySeq01"] = "cag cta".replace(" ","").upper() # x
# dictTargetSequence["mySeq02"] = "cac ata".replace(" ","").upper() # y
# scoringWeight = [1, -1, -1]
dictTargetSequence["mySeq01"] = "atg ctg g".replace(" ","").upper() # x
dictTargetSequence["mySeq02"] = "atg GAg A".replace(" ","").upper() # y
scoringWeight = [2, -2, -1]
# scoreMatrix = np.zeros((len(dictTargetSequence["mySeq02"]) + 1, len(dictTargetSequence["mySeq01"]) + 1))
w, h = len(dictTargetSequence["mySeq01"]) + 3, len(dictTargetSequence["mySeq02"]) + 3
scoreMatrixAllValues = [[[] for x in range(1,w)] for y in range(1,h)]
scoreMatrixMaxValues = [[[] for x in range(1,w)] for y in range(1,h)]
scoreMatrixArrows = [[[] for x in range(1,w)] for y in range(1,h)]
scoreMatrixTraceback = [[[] for x in range(1,w)] for y in range(1,h)]
for x in range(2, len(dictTargetSequence["mySeq01"]) + 2):
scoreMatrixAllValues[0][x] = dictTargetSequence["mySeq01"][x - 2]
scoreMatrixMaxValues[0][x] = dictTargetSequence["mySeq01"][x - 2]
scoreMatrixArrows[0][x] = dictTargetSequence["mySeq01"][x - 2]
scoreMatrixTraceback[0][x] = dictTargetSequence["mySeq01"][x - 2]
for y in range(2, len(dictTargetSequence["mySeq02"]) + 2):
scoreMatrixAllValues[y][0] = dictTargetSequence["mySeq02"][y - 2]
scoreMatrixMaxValues[y][0] = dictTargetSequence["mySeq02"][y - 2]
scoreMatrixArrows[y][0] = dictTargetSequence["mySeq02"][y - 2]
scoreMatrixTraceback[y][0] = dictTargetSequence["mySeq02"][y - 2]
scoreMatrixAllValues[1][1] = 0
scoreMatrixMaxValues[1][1] = 0
for x in range(1, len(dictTargetSequence["mySeq01"]) + 2):
scoreMatrixAllValues[1][x] = int((x - 1) * scoringWeight[2])
scoreMatrixMaxValues[1][x] = int((x - 1) * scoringWeight[2])
scoreMatrixArrows[1][x] = [0]
for y in range(1, len(dictTargetSequence["mySeq02"]) + 2):
scoreMatrixAllValues[y][1] = int((y - 1) * scoringWeight[2])
scoreMatrixMaxValues[y][1] = int((y - 1) * scoringWeight[2])
scoreMatrixArrows[y][1] = [2]
scoreMatrixArrows[1][1] = "-"
scoreMatrixTraceback[1][1] = "-"
# determine score matrix
for jj in range(2,len(dictTargetSequence["mySeq02"]) + 2): # y-axis
for ii in range(2,len(dictTargetSequence["mySeq01"]) + 2): # x-axis
# if ii > 0 and jj > 0:
T = []
if dictTargetSequence["mySeq02"][jj - 2] == dictTargetSequence["mySeq01"][ii - 2]:
scoreTmp = scoringWeight[0]
elif dictTargetSequence["mySeq02"][jj - 2] != dictTargetSequence["mySeq01"][ii - 2]:
scoreTmp = scoringWeight[1]
T.append(scoreMatrixMaxValues[jj][ii-1] + scoringWeight[2]) # left
T.append(scoreMatrixMaxValues[jj-1][ii-1] + scoreTmp) # diagonal
T.append(scoreMatrixMaxValues[jj-1][ii] + scoringWeight[2]) # up
scoreMatrixAllValues[jj][ii] = T
scoreMatrixMaxValues[jj][ii] = max(T)
scoreMatrixArrows[jj][ii] = [i for i, j in enumerate(T) if j == max(T)]
dashlineLength = 140
print("-" * dashlineLength)
print(" " * 40 + "Score matrix with all value-triplets".upper())
print(" " * 43 + "(0: left, 1: diagonal, 2: up)".upper())
print("-" * dashlineLength)
# print("\n")
for jj in range(len(scoreMatrixAllValues)):
print("\t", end='')
for ii in range(len(scoreMatrixAllValues[jj])):
currentValue = scoreMatrixAllValues[jj][ii]
if isinstance(currentValue, list):
print(currentValue, "\t", end='')
if isinstance(currentValue, str):
if jj == 0:
print(currentValue, (15 - len(currentValue)) * " ", end = '')
else:
print(currentValue, "\t", end = '')
if isinstance(currentValue, int):
if jj == 0:
print(currentValue, (5-len(str(currentValue))) * " ", end = '')
elif jj == 1 and ii > 1:
print(currentValue, "\t\t", end='')
else:
print(currentValue, "\t", end = '')
if jj < (len(scoreMatrixAllValues) - 1):
print("\n")
print("")
print("-" * dashlineLength)
print("\n")
print("-" * dashlineLength)
print(" " * 40 + "Score matrix with highest values".upper())
print("-" * dashlineLength)
# print("\n")
for jj in range(len(scoreMatrixMaxValues)):
print("\t\t\t\t", end = '')
for ii in range(len(scoreMatrixMaxValues[jj])):
currentValue = scoreMatrixMaxValues[jj][ii]
if isinstance(currentValue, list):
print(currentValue, "\t", end = '')
if isinstance(currentValue, str):
if jj == 0:
print(currentValue, "\t", end = '')
else:
print(currentValue, "\t", end = '')
if isinstance(currentValue, int):
if jj == 0:
print(currentValue, (5 - len(str(currentValue))) * " ", end = '')
elif jj == 1 and ii > 1:
print(currentValue, "\t", end = '')
else:
print(currentValue, "\t", end = '')
if jj < (len(scoreMatrixMaxValues) - 1):
print("\n")
print("")
print("-" * dashlineLength)
print("\n")
print("-" * dashlineLength)
print(" " * 40 + "Score martrix showing the Arrowhead Orientation".upper())
print(" " * 43 + "(0: left, 1: diagonal, 2: up)".upper())
print("-" * dashlineLength)
# print("\n")
for jj in range(len(scoreMatrixArrows)):
print("\t\t\t\t", end='')
for ii in range(len(scoreMatrixArrows[jj])):
currentValue = scoreMatrixArrows[jj][ii]
if isinstance(currentValue, list):
print(currentValue, "\t", end='')
if isinstance(currentValue, str):
if jj == 0:
print(currentValue, "\t", end='')
else:
print(currentValue, "\t", end='')
if isinstance(currentValue, int):
if jj == 0:
print(currentValue, (5-len(str(currentValue))) * " ", end='')
elif jj == 1 and ii>1:
print(currentValue, "\t", end = '')
else:
print(currentValue, "\t", end = '')
if jj < (len(scoreMatrixArrows) - 1):
print("\n")
print("")
print("-" * dashlineLength)
print("\n")
--------------------------------------------------------------------------------------------------------------------------------------------
SCORE MATRIX WITH ALL VALUE-TRIPLETS
(0: LEFT, 1: DIAGONAL, 2: UP)
--------------------------------------------------------------------------------------------------------------------------------------------
[] [] A T G C T G G
[] 0 -1 -2 -3 -4 -5 -6 -7
A -1 [-2, 2, -2] [1, -3, -3] [0, -4, -4] [-1, -5, -5] [-2, -6, -6] [-3, -7, -7] [-4, -8, -8]
T -2 [-3, -3, 1] [0, 4, 0] [3, -1, -1] [2, -2, -2] [1, 1, -3] [0, -4, -4] [-1, -5, -5]
G -3 [-4, -4, 0] [-1, -1, 3] [2, 6, 2] [5, 1, 1] [4, 0, 0] [3, 3, -1] [2, 2, -2]
G -4 [-5, -5, -1] [-2, -2, 2] [1, 5, 5] [4, 4, 4] [3, 3, 3] [2, 6, 2] [5, 5, 1]
A -5 [-6, -2, -2] [-3, -3, 1] [0, 0, 4] [3, 3, 3] [2, 2, 2] [1, 1, 5] [4, 4, 4]
G -6 [-7, -7, -3] [-4, -4, 0] [-1, 3, 3] [2, 2, 2] [1, 1, 1] [0, 4, 4] [3, 7, 3]
A -7 [-8, -4, -4] [-5, -5, -1] [-2, -2, 2] [1, 1, 1] [0, 0, 0] [-1, -1, 3] [2, 2, 6]
--------------------------------------------------------------------------------------------------------------------------------------------
--------------------------------------------------------------------------------------------------------------------------------------------
SCORE MATRIX WITH HIGHEST VALUES
--------------------------------------------------------------------------------------------------------------------------------------------
[] [] A T G C T G G
[] 0 -1 -2 -3 -4 -5 -6 -7
A -1 2 1 0 -1 -2 -3 -4
T -2 1 4 3 2 1 0 -1
G -3 0 3 6 5 4 3 2
G -4 -1 2 5 4 3 6 5
A -5 -2 1 4 3 2 5 4
G -6 -3 0 3 2 1 4 7
A -7 -4 -1 2 1 0 3 6
--------------------------------------------------------------------------------------------------------------------------------------------
--------------------------------------------------------------------------------------------------------------------------------------------
SCORE MARTRIX SHOWING THE ARROWHEAD ORIENTATION
(0: LEFT, 1: DIAGONAL, 2: UP)
--------------------------------------------------------------------------------------------------------------------------------------------
[] [] A T G C T G G
[] - [0] [0] [0] [0] [0] [0] [0]
A [2] [1] [0] [0] [0] [0] [0] [0]
T [2] [2] [1] [0] [0] [0, 1] [0] [0]
G [2] [2] [2] [1] [0] [0] [0, 1] [0, 1]
G [2] [2] [2] [1, 2] [0, 1, 2] [0, 1, 2] [1] [0, 1]
A [2] [1, 2] [2] [2] [0, 1, 2] [0, 1, 2] [2] [0, 1, 2]
G [2] [2] [2] [1, 2] [0, 1, 2] [0, 1, 2] [1, 2] [1]
A [2] [1, 2] [2] [2] [0, 1, 2] [0, 1, 2] [2] [2]
--------------------------------------------------------------------------------------------------------------------------------------------
# determine traceback path
xpos = w - 2
ypos = h - 2
scoreMatrixTraceback[ypos][xpos] = 1
pathList = []
while xpos > 0 and ypos > 0:
# print(ypos, xpos)
# print(scoreMatrixArrows[ypos][xpos])
if isinstance(scoreMatrixArrows[ypos][xpos], list):
if 1 in scoreMatrixArrows[ypos][xpos]:
scoreMatrixTraceback[ypos][xpos] = 1
xpos = xpos - 1
ypos = ypos - 1
pathList.append("D")
elif 0 in scoreMatrixArrows[ypos][xpos]:
scoreMatrixTraceback[ypos][xpos] = 1
xpos = xpos - 1
ypos = ypos
pathList.append("L")
elif 2 in scoreMatrixArrows[ypos][xpos]:
scoreMatrixTraceback[ypos][xpos] = 1
xpos = xpos
ypos = ypos - 1
pathList.append("U")
elif scoreMatrixArrows[ypos][xpos] == "-":
scoreMatrixTraceback[ypos][xpos] = 1
xpos = 0
ypos = 0
if xpos != 0 and ypos != 0:
print("ERROR: starting position not reached")
print("-" * dashlineLength)
print(" " * 40 + "Score matrix with highest values")
print(" " * 43 + "(0: left, 1: diagonal, 2: up)")
print("-" * dashlineLength)
for jj in range(len(scoreMatrixTraceback)):
print("\t\t\t\t", end='')
for ii in range(len(scoreMatrixTraceback[jj])):
currentValue = scoreMatrixTraceback[jj][ii]
if isinstance(currentValue, list):
print(currentValue, "\t", end='')
if isinstance(currentValue, str):
if jj == 0:
print(currentValue, "\t", end='')
else:
print(currentValue, "\t", end='')
if isinstance(currentValue, int):
if jj == 0:
print(currentValue, (5-len(str(currentValue))) * " ", end='')
elif jj == 1 and ii>1:
print(currentValue, "\t", end='')
else:
print(currentValue, "\t", end='')
if jj < (len(scoreMatrixTraceback)-1):
print("\n")
print("")
print("-" * dashlineLength)
print("\n")
--------------------------------------------------------------------------------------------------------------------------------------------
Score matrix with highest values
(0: left, 1: diagonal, 2: up)
--------------------------------------------------------------------------------------------------------------------------------------------
[] [] A T G C T G G
[] 1 [] [] [] [] [] [] []
A [] 1 [] [] [] [] [] []
T [] [] 1 [] [] [] [] []
G [] [] [] 1 1 1 [] []
G [] [] [] [] [] [] 1 []
A [] [] [] [] [] [] 1 []
G [] [] [] [] [] [] [] 1
A [] [] [] [] [] [] [] 1
--------------------------------------------------------------------------------------------------------------------------------------------
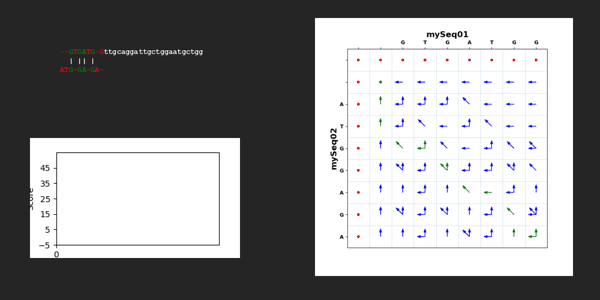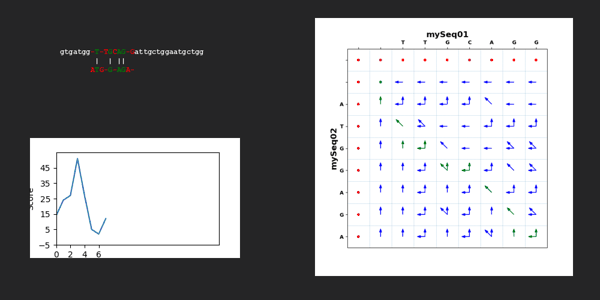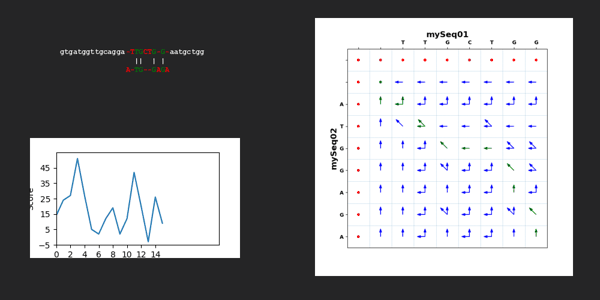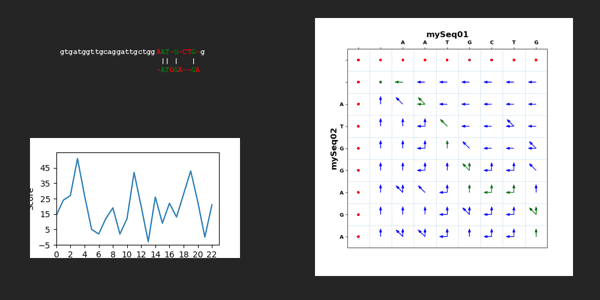
# https://stackoverflow.com/questions/72751096/plot-a-grid-with-arrows-on-it
import numpy as np
import matplotlib.pyplot as plt
import matplotlib
mySeq02Reverse = list(reversed(dictTargetSequence["mySeq02"]))
scale = 0.2
fig, ax = plt.subplots(figsize=(7, 7))
ax.set_xlim(-0.5, len(["",*dictTargetSequence["mySeq01"]]) + 0.5)
ax.set_ylim(-0.5, len(["",*dictTargetSequence["mySeq02"]]) + 0.5)
ax.set_xticks(range(len(["",*dictTargetSequence["mySeq01"]])+1),["","",*dictTargetSequence["mySeq01"]], weight = 'bold')
ax.set_yticks(range(len([*mySeq02Reverse])+2),[*mySeq02Reverse,"",""], weight = 'bold')
ax.tick_params(top=True, labeltop=True, bottom=False, labelbottom=False)
ax.set_title('mySeq01', fontsize=16, weight = 'bold')
ax.set_ylabel('mySeq02', fontsize=16, weight = 'bold')
ii=0.1
jj=0.1
# plt.text(ii,jj, "x", horizontalalignment='center', verticalalignment='center', transform=ax.transAxes)
for jj in range(0,len(dictTargetSequence["mySeq02"]) + 2): # y-axis
plt.axhline(y = jj - 0.5, linestyle = '--', alpha = 0.5,linewidth = 0.5)
for ii in range(0,len(dictTargetSequence["mySeq01"]) + 2): # x-axis
if jj == 0:
plt.axvline(x = ii-0.5, linestyle = '--', alpha = 0.5,linewidth = 0.5)
if ii > 1 and jj > 0:
if scoreMatrixTraceback[jj][ii] == 1:
arrowcolor="green"
else:
arrowcolor="blue"
if 1 in scoreMatrixArrows[jj][ii]:
plt.arrow(ii,len(dictTargetSequence["mySeq02"]) + 2 - jj-1, scale*(-1), scale*(1), head_width=0.1, color=arrowcolor)
if 0 in scoreMatrixArrows[jj][ii]:
plt.arrow(ii,len(dictTargetSequence["mySeq02"]) + 2 - jj-1, scale*(-1), scale*(0), head_width=0.1, color=arrowcolor)
if 2 in scoreMatrixArrows[jj][ii]:
plt.arrow(ii,len(dictTargetSequence["mySeq02"]) + 2 - jj-1, scale*(0), scale*(1), head_width=0.1, color=arrowcolor)
if ii==1 and jj > 1:
if scoreMatrixTraceback[jj][ii] == 1:
arrowcolor="green"
else:
arrowcolor="blue"
if 1 in scoreMatrixArrows[jj][ii]:
plt.arrow(ii,len(dictTargetSequence["mySeq02"]) + 2 - jj - 1, scale*(-1), scale*(1), head_width=0.1, color=arrowcolor)
if 0 in scoreMatrixArrows[jj][ii]:
plt.arrow(ii,len(dictTargetSequence["mySeq02"]) + 2 - jj - 1, scale*(-1), scale*(0), head_width=0.1, color=arrowcolor)
if 2 in scoreMatrixArrows[jj][ii]:
plt.arrow(ii,len(dictTargetSequence["mySeq02"]) + 2 - jj - 1, scale*(0), scale*(1), head_width=0.1, color=arrowcolor)
if jj==1 and ii==1:
# plt.Circle((ii,len(dictTargetSequence["mySeq02"]) + 2 - jj-1), 0.2, color='r')
circle1 = plt.Circle((ii,len(dictTargetSequence["mySeq02"]) + 2 - jj-1), 0.05, color='g')
ax.add_patch(circle1)
if jj < 1 or ii < 1:
circle1 = plt.Circle((ii,len(dictTargetSequence["mySeq02"]) + 2 - jj-1), 0.05, color='r')
ax.add_patch(circle1)
if saveGallery == True:
plt.savefig('./../GALLERY/Sequence Analysis/03_02_Needleman_Wunsch_Algorithm.png', bbox_inches='tight')
plt.show()
dashlineLength = 40
indentSpace = 3
charSpacing = 3
print("-" * dashlineLength)
print(" " * 7 + "PATH-RESULT DERIVED FROM PLOT")
print(" " * 7 + "(L: left, D: diagonal, U: up)")
print("-" * dashlineLength)
print(" "*indentSpace,end='')
for ii in range(len(pathList)):
print(pathList[ii],end='')
print(" " * charSpacing, end = '')
print("")
print("-" * dashlineLength)
pathListReverse = list(reversed(pathList))

----------------------------------------
PATH-RESULT DERIVED FROM PLOT
(L: left, D: diagonal, U: up)
----------------------------------------
U D U D L L D D D
----------------------------------------
alignedTargetSequence = {}
alignedTargetSequence["mySeq01"] = []
alignedTargetSequence["mySeq02"] = []
mySeq01Pos = 0
mySeq02Pos = 0
for ii in range(len(pathList)):
if pathListReverse[ii] == "D":
alignedTargetSequence["mySeq01"].append(dictTargetSequence["mySeq01"][mySeq01Pos])
alignedTargetSequence["mySeq02"].append(dictTargetSequence["mySeq02"][mySeq02Pos])
mySeq01Pos += 1
mySeq02Pos += 1
elif pathListReverse[ii] == "L":
alignedTargetSequence["mySeq01"].append(dictTargetSequence["mySeq01"][mySeq01Pos])
alignedTargetSequence["mySeq02"].append("-")
mySeq01Pos += 1
# mySeq02Pos += 1
elif pathListReverse[ii] == "U":
alignedTargetSequence["mySeq01"].append("-")
alignedTargetSequence["mySeq02"].append(dictTargetSequence["mySeq02"][mySeq02Pos])
# mySeq01Pos += 1
mySeq02Pos += 1
dashlineLength = 70
indentSpace = 20
charSpacing = 3
print("-" * dashlineLength)
print(" " * 7 + "ALIGNMENT-RESULT BASED ON NEEDLEMAN-WUNSCH-ALGORITHM")
print("-" * dashlineLength)
print(" mySeq01: ",end='')
print(" "*(indentSpace-len(" mySeq01: ")),end='')
for ii in range(len(alignedTargetSequence["mySeq01"])):
print(alignedTargetSequence["mySeq01"][ii],end='')
print(" " * charSpacing, end = '')
print("")
print(" "*indentSpace,end='')
for ii in range(len(alignedTargetSequence["mySeq02"])):
if alignedTargetSequence["mySeq01"][ii] == alignedTargetSequence["mySeq02"][ii]:
print("|",end='')
else:
print(" ",end='')
print(" " * charSpacing, end = '')
print("")
print(" mySeq02: ",end='')
print(" "*(indentSpace-len(" mySeq02: ")),end='')
for ii in range(len(alignedTargetSequence["mySeq02"])):
print(alignedTargetSequence["mySeq02"][ii],end='')
print(" " * charSpacing, end = '')
print("")
print("-" * dashlineLength)
print("\n")
----------------------------------------------------------------------
ALIGNMENT-RESULT BASED ON NEEDLEMAN-WUNSCH-ALGORITHM
----------------------------------------------------------------------
mySeq01: A T G C T G - G -
| | | | |
mySeq02: A T G - - G A G A
----------------------------------------------------------------------
%%capture
# https://www.blog.pythonlibrary.org/2021/02/02/drawing-text-on-images-with-pillow-and-python/
import ModulesOwn.A_Groundwork as Groundwork
import ModulesOwn.C_Mutations as Mutations
from IPython.display import clear_output
import time
import PIL
from ModulesExternal.TerminalColors import TerminalColors
import matplotlib
import matplotlib.pyplot as plt
import numpy as np
from PIL import Image, ImageDraw, ImageFont
# matplotlib.use('Agg')
# https://thesmithfam.org/blog/2012/10/25/temporarily-suppress-console-output-in-python/
from contextlib import contextmanager
import sys, os
colorRed = "\033[31m"
colorGreen = "\033[32m"
colorDefault = "\033[39m"
cgreen = [0,255,0]
cred = [255,0,0]
cdefaucgreen= [0,0,0]
alignedTargetSequence={}
scoreAllWindow=[]
dictTargetSequence = {}
dictTargetSequence["mySeq01"] = "Gtg Atg gTt gcA gga Ttg ctg gaA tgc tgg".replace(" ","").upper() # x
dictTargetSequence["mySeq02"] = "atg GAg A".replace(" ","").upper() # y
scoringWeight = [2, -5, -1]
mySeq01StrColor=""
mySeq02StrColor=""
indentCount = 20
for kk in range(len(dictTargetSequence["mySeq01"])-len(dictTargetSequence["mySeq02"])+1):
scoreAll=0
scoreMatrixAllValues, scoreMatrixMaxValues, scoreMatrixArrows = Mutations.needlemanWunschAlgorithm(dictTargetSequence["mySeq01"][kk:kk+len(dictTargetSequence["mySeq02"])], dictTargetSequence["mySeq02"], scoringWeight)
scoreMatrixTraceback, pathList = Mutations.needlemanWunschTracebackPathList(dictTargetSequence["mySeq01"][kk:kk+len(dictTargetSequence["mySeq02"])], dictTargetSequence["mySeq02"], scoreMatrixArrows)
alignedTargetSequence["mySeq01"], alignedTargetSequence["mySeq02"] = Mutations.needlemanWunschComputeResult(dictTargetSequence["mySeq01"][kk:kk+len(dictTargetSequence["mySeq02"])], dictTargetSequence["mySeq02"], pathList)
for jj in range(2,len(scoreMatrixTraceback)):
for ii in range(2,len(scoreMatrixTraceback[jj])):
if scoreMatrixTraceback[jj][ii] == 1:
scoreAll = scoreAll + scoreMatrixTraceback[jj][ii] * scoreMatrixMaxValues[jj][ii]
scoreAllWindow.append(scoreAll)
mySeq01Str = ''.join(alignedTargetSequence["mySeq01"])
mySeq02Str = ''.join(alignedTargetSequence["mySeq02"])
mySeq01StrBeginning = dictTargetSequence["mySeq01"][:kk].lower()
mySeq01StrEnd = dictTargetSequence["mySeq01"][kk+len(dictTargetSequence["mySeq02"]):].lower()
xpos = 30
ypos = 30
image = Mutations.text_color(xpos, ypos, mySeq01StrBeginning, mySeq01Str, mySeq01StrEnd, mySeq02Str, scoreAllWindow, scoreMatrixTraceback, scoreMatrixArrows, dictTargetSequence["mySeq01"], dictTargetSequence["mySeq02"],kk)
image.save("./../NeedlemanWunschSeq/"+str(f"{kk:02d}")+".png")
time.sleep(0.1)
import numpy as np
from PIL import Image
from PIL import Image
import math
import imageio
import random
import os
frames_All=[]
for kk in range(23):
img = Image.open("./../NeedlemanWunschSeq/"+str(f"{kk:02d}")+".png")
img=img.resize((600,300))
# print("hello")
frames_All.append(img)
frames_All[0].save('./gifs/NeedlemanWunschSeq.gif',
save_all = True, append_images = frames_All[:],
optimize = True, loop = 0, duration = 420, format='GIF', disposal=2)
# 2BeWorkedOn
This section is about the analysis of a target sequence in view of it's k-mer subcomponents. It is a direct preparation to put together small DNA-fragments with overlap in order to get the full picture, as it is required for NGS-data exploration of very large DNA sequences.
# data sources
# RNA-Sequence: S. cerevisiae initiator methionyl tRNAs
# Kolitz SE, Lorsch JR. Eukaryotic initiator tRNA: finely tuned and ready for action.
# FEBS Lett. 2010 Jan 21;584(2):396-404. doi: 10.1016/j.febslet.2009.11.047.
# PMID: 19925799; PMCID: PMC2795131.
# - https://pubmed.ncbi.nlm.nih.gov/19925799/
#
# Tertiary structure scheme of t-RNA published by user Yikrazuul under
# the Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0) license
# - https://en.wikipedia.org/wiki/Transfer_RNA#/media/File:TRNA-Phe_yeast_1ehz.png
# adapted in terms of labeling
#
# https://www.ncbi.nlm.nih.gov/nuccore/S70505.1
# AGCAGAGUGGCGCAGCGGAAGCGCGCUGGGCCCAUAACCCAGAGGDCGAZGGAUCGAAACCAUCCUCUFCUACCA
# dictTargetSequence = {}
# - https://de.wikipedia.org/wiki/TRNA
# include modules
from Bio.Seq import Seq
from IPython.display import Image as ImageIPython
import json
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
import ModulesOwn.D_KmerAnalysis as KmerAnalysis
import os
from sty import fg, bg, ef, rs
# initialize and define variables and parameter
# Acceptor stem: 1 and 6
# D arm : 2
# Anticodon arm: 3
# Variable loop: 4
# T arm : 5
# CCA tail : 7
# 1 2 3 4 5 6 7
Seq_tmp0 = "AGCGCCGU GGCGCAGDGGAAGCGC GCAGGGCUCAUAACCCUG AUGDC CUCGGAUCGAAACCGAG CGGCGCU ACCA"
Seq_tmp = Seq_tmp0.replace(" ","")
dictSeqGroup = {"targetSeq": {
"mySequence": Seq_tmp,
}
}
myColors = [TerminalColors.Green, TerminalColors.Blue, TerminalColors.Yellow]
myDefaultColor = TerminalColors.Default
myColorWarning = TerminalColors.Red
# load data
if os.path.exists("./Part01Data/04_01_DictSeqGroup.json"):
os.remove("./Part01Data/04_01_DictSeqGroup.json")
# compute and visualize results
sequence_colored = fg(128,0,200) + "AGCGCCGU" + fg(128,0,0) + "GGCGCAGDGGAAGCGC" + fg(0,94,254) + "GCAGGGCU" + fg(100,100,100)
sequence_colored = sequence_colored + "CAU" + fg(0,94,254) + "AACCCUG" + fg(211,85,0) + "AUGDC" + fg(32,118,0) + "CUCGGAUCGAAACCGAG" + fg(128,0,200)
sequence_colored = sequence_colored + "CGGCGCU" + f"{TerminalColors.Yellow}" + "ACCA" + f"{TerminalColors.Default}"
print("-" * 99, end='\n')
print("\t\t\tTertiary structure scheme of a tRNA molecule", end='\n')
print("-" * 99, end='\n')
print("Acceptor stem: \t " + fg(128,0,200) + "purple \n" + f"{TerminalColors.Default}" +
"D arm: \t\t " + fg(128,0,0) + "red \n" + f"{TerminalColors.Default}" +
"Anticodon arm: \t " + fg(0,94,254) + "blue \n" + f"{TerminalColors.Default}" +
"Anticodon: \t " + fg(100,100,100) + "black \n" + f"{TerminalColors.Default}" +
"CCA tail: \t " + f"{TerminalColors.Yellow}" + "yellow \n" + f"{TerminalColors.Default}" +
"Variable loop: \t " + fg(211,85,0) + "orange \n" + f"{TerminalColors.Default}" +
"T arm: \t\t " + fg(32,118,0) + "green" + f"{TerminalColors.Default}")
print("")
print("-" * 99, end='\n')
print("Target Sequence: \t" + sequence_colored)
print("RNA-Sequence: S. cerevisiae initiator methionyl tRNAs")
print("Source: Kolitz SE, Lorsch JR. Eukaryotic initiator tRNA: finely tuned and ready for action.")
print("FEBS Lett. 2010 Jan 21;584(2):396-404. doi: 10.1016/j.febslet.2009.11.047.")
print("PMID: 19925799; PMCID: PMC2795131.")
print("https://pubmed.ncbi.nlm.nih.gov/19925799/")
print("-" * 99, end='\n')
display(ImageIPython(filename = './Figures_scientific/TRNA-Phe_yeast.png', width=300))
print("Tertiary structure scheme of t-RNA published by user Yikrazuul under")
print("the Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0) license.")
print("https://en.wikipedia.org/wiki/Transfer_RNA#/media/File:TRNA-Phe_yeast_1ehz.png")
print("")
print("-" * 99, end='\n')
print("\t\t\t\tPalindroms as identified by algorithm")
print("-" * 99, end='\n')
print(f"{myDefaultColor}" + "Forward Sequence: \t " + f"{myColors[0]}" + "green" + f"{myDefaultColor}")
print("Reverse Complement: \t " + f"{myColors[1]}" + "blue" + f"{myDefaultColor}")
print("Overlays: \t\t " + f"{myColors[2]}" + "yellow" + f"{myDefaultColor}")
print("")
print("KLENGTH = 5")
print("-" * 99, end='\n')
klength = 5
dictSeqGroup = KmerAnalysis.FindPalindroms(dictSeqGroup, klength)
print("Target Sequence: \t" + sequence_colored)
print("-" * 99, end='\n')
with open("./ModulesOwn/D_KmerAnalysis/04_01_DictSeqGroup.json", "w") as outfile:
json.dump(dictSeqGroup, outfile, indent = 4, sort_keys = True)
file1 = open('./ModulesOwn/D_KmerAnalysis/04_01_DictSeqGroup.json', 'r')
Lines = file1.readlines()
count = 0
for line in Lines:
count += 1
file1.close()
seqstr=Groundwork.List2String(dictSeqGroup["targetSeq"]["mySequence"])
stringlist = KmerAnalysis.PrintPalindroms(dictSeqGroup, klength, myColors, myDefaultColor)
for ii in range(len(stringlist)):
print("Palindroms "+"{:02d}".format(ii+1) + ": \t\t" +stringlist[ii])
print("")
print("KLENGTH = 7")
klength = 7
dictSeqGroup = KmerAnalysis.FindPalindroms(dictSeqGroup, klength)
print("-" * 99, end='\n')
print("Target Sequence: \t" + sequence_colored)
print("-" * 99, end='\n')
with open("./ModulesOwn/D_KmerAnalysis/04_01_DictSeqGroup.json", "w") as outfile:
json.dump(dictSeqGroup, outfile, indent = 4, sort_keys = True)
file1 = open('./ModulesOwn/D_KmerAnalysis/04_01_DictSeqGroup.json', 'r')
Lines = file1.readlines()
count = 0
for line in Lines:
count += 1
file1.close()
seqstr = Groundwork.List2String(dictSeqGroup["targetSeq"]["mySequence"])
stringlist = KmerAnalysis.PrintPalindroms(dictSeqGroup, klength, myColors, myDefaultColor)
for ii in range(len(stringlist)):
print("Palindroms "+"{:02d}".format(ii+1) + ": \t\t" +stringlist[ii])
--------------------------------------------------------------------------------------------------- Tertiary structure scheme of a tRNA molecule --------------------------------------------------------------------------------------------------- Acceptor stem: purple D arm: red Anticodon arm: blue Anticodon: black CCA tail: yellow Variable loop: orange T arm: green --------------------------------------------------------------------------------------------------- Target Sequence: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA RNA-Sequence: S. cerevisiae initiator methionyl tRNAs Source: Kolitz SE, Lorsch JR. Eukaryotic initiator tRNA: finely tuned and ready for action. FEBS Lett. 2010 Jan 21;584(2):396-404. doi: 10.1016/j.febslet.2009.11.047. PMID: 19925799; PMCID: PMC2795131. https://pubmed.ncbi.nlm.nih.gov/19925799/ ---------------------------------------------------------------------------------------------------

Tertiary structure scheme of t-RNA published by user Yikrazuul under the Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0) license. https://en.wikipedia.org/wiki/Transfer_RNA#/media/File:TRNA-Phe_yeast_1ehz.png --------------------------------------------------------------------------------------------------- Palindroms as identified by algorithm --------------------------------------------------------------------------------------------------- Forward Sequence: green Reverse Complement: blue Overlays: yellow KLENGTH = 5 --------------------------------------------------------------------------------------------------- Target Sequence: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA --------------------------------------------------------------------------------------------------- Palindroms 01: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 02: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 03: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 04: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 05: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 06: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 07: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA KLENGTH = 7 --------------------------------------------------------------------------------------------------- Target Sequence: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA --------------------------------------------------------------------------------------------------- Palindroms 01: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 02: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 03: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 04: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 05: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 06: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA Palindroms 07: AGCGCCGUGGCGCAGDGGAAGCGCGCAGGGCUCAUAACCCUGAUGDCCUCGGAUCGAAACCGAGCGGCGCUACCA
Goal: To visualize the quality scores of a short FastQ sequence.
# sources
# - https://www.biostars.org/p/317524/
# - https://gatk.broadinstitute.org/hc/en-us/articles/360035531872-Phred-scaled-quality-scores
# - https://www.geeksforgeeks.org/plot-a-vertical-line-in-matplotlib/
# - https://en.wikipedia.org/wiki/FASTQ_format
# - https://stackoverflow.com/questions/21465988/python-equivalent-to-hold-on-in-matlab
# - https://stackoverflow.com/questions/63470319/pyplot-add-single-text-to-xaxis-like-a-tick
# data sources
# Target Sequence taken from the example of the wikipedia article:
# - https://en.wikipedia.org/wiki/FASTQ_format
# include modules
import matplotlib.pyplot as plt
from ModulesExternal.TerminalColors import TerminalColors
import ModulesOwn.A_Groundwork as Groundwork
import numpy as np
# initialize and define variables and parameter
myDefaultColor = TerminalColors.Default
dictTargetSequence = {}
dictTargetSequence["mySequence"] = "GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAAT"
dictTargetSequence["Quality"] = "!''*((((***+))%%%++)(%%%%).1***-+*''))"
labelNum, labelX = Groundwork.SeqNumberGen(dictTargetSequence["mySequence"], 10)
# compute and visualize results
def phredQualityTranform(mySeqQuality):
plotvalues = []
for ii in range(len(mySeqQuality)):
plotvalues.append(ord(mySeqQuality[ii]))
return plotvalues
plotvalues = phredQualityTranform(dictTargetSequence["Quality"])
x = np.arange(0, len(dictTargetSequence["Quality"]))
mcolor=[]
for ii in range(len(x)-1):
if dictTargetSequence["mySequence"][ii] == "G":
mcolor.append(TerminalColors.Green)
elif dictTargetSequence["mySequence"][ii] == "C":
mcolor.append(TerminalColors.Red)
else:
mcolor.append(TerminalColors.Blue)
plt.figure(figsize=(12,5))
ax = plt.axes()
plt.axis([0, len(plotvalues), min(plotvalues), max(plotvalues)])
for ii in range(len(x) - 1):
if plotvalues[ii] < plotvalues[ii + 1]:
mcolor="green"
elif plotvalues[ii] > plotvalues[ii + 1]:
mcolor="red"
else:
mcolor="blue"
plt.plot(x[ii:ii + 2],plotvalues[ii:ii + 2], color = mcolor, linestyle = 'dashed', linewidth = 3) #, marker='o', markerfacecolor = mcolor, markersize = 10
plt.text(ii - 0.25, min(plotvalues) - 1, dictTargetSequence["mySequence"][ii], color = "black", weight = "bold",fontsize = 12)
plt.axvline(x = ii + 1, color = "black", linestyle='dashed', linewidth = 0.2, label = 'axvline - full height')
plt.text(ii + 0.75, min(plotvalues) - 1, dictTargetSequence["mySequence"][ii + 1], color = "black", weight = "bold",fontsize = 12)
plt.tick_params(axis = 'x', which = 'both', bottom = False, top = False, labelbottom = False)
plt.title("FastQ", weight = "bold", fontsize = 24)
plt.ylabel('Phred Score',weight="bold",fontsize = 16)
plt.xlim(0, max(x))
plt.xticks(x, dictTargetSequence["mySequence"])
plt.yticks(np.arange(min(plotvalues), max(plotvalues) + 1, 1.0), weight = "bold", fontsize = 12)
plt.ylim(min(plotvalues), max(plotvalues))
if saveGallery == True:
plt.savefig('./../GALLERY/Sequence Analysis/05_01_Phred_Quality_Score.png', bbox_inches='tight')
plt.show()

# export code to html
import os
os.system("jupyter nbconvert SequenceAnalysis.ipynb --to html")
fin = open("SequenceAnalysis.html", "rt", encoding="utf8")
fout = open("SequenceAnalysisPart01_mod.html", "wt", encoding="utf8")
for line in fin:
if line.find("<ol>") > -1:
line = line.replace("<ol>", "<ol type=\"1\">")
elif line.find("<li>") > -1:
line = line.replace("<li>", "<li type=\"1\">")
fout.write(line)
fin.close()
fout.close()
import os
if os.path.isfile("SequenceAnalysis.html"):
os.remove("SequenceAnalysis.html")
os.rename("SequenceAnalysisPart01_mod.html", "SequenceAnalysis.html")